
Plaintiffs in Supreme Court abortion pill case say FDA is ‘jeopardizing women’s health’
By Carole Novielli, originally published in LiveAction on February 26, 2024
A group of pro-life doctors who are plaintiffs in an upcoming U.S. Supreme Court abortion pill case have filed their brief seeking to hold the Food and Drug Administration (FDA) accountable against an alleged “unfettered power.” The plaintiffs believe the FDA failed to protect women when they moved to expand use of the approved abortion drug, mifepristone. The case is set to be heard on March 26, 2024.
The brief, filed in Alliance for Hippocratic Medicine (AHM) v. Food and Drug Administration (FDA) claimed that the “FDA’s removal of safeguards for abortion drugs was arbitrary, capricious, an abuse of discretion, and otherwise unlawful,” the pro-life doctors and medical organizations claimed in their brief.
“FDA’s precipitous decision to remove its ‘longstanding, minimally burdensome’ in-office visit…was a textbook violation of the APA [Administrative Procedure Act],” AHM wrote.
History of Abortion Pill Approval and Conflicts
AHM originally sought to question the legitimacy of the 2000 approval of the abortion pill by the FDA. However, due to previous decisions in the case, arguments before the High Court will center around changes made by the FDA to the drug’s REMS safety requirements in 2016 and 2021.
Mifepristone was approved by the FDA in 2000 to be prescribed in-person through the 7th week of pregnancy. In 2016, the FDA removed requirements that all non-fatal adverse events be reported. It also allowed the pill to be prescribed through the 10th week of pregnancy, removed the requirement that only physicians prescribe the drug, and no longer required women to take the first pill at the abortion facility.
In 2021, the Biden FDA eliminated the in-person dispensing requirement and allowed for the abortion pill to be permanently shipped by mail. Then in 2023, the FDA further expanded access to allow retail pharmacies to dispense the drug.
Live Action News previously documented potential conflicts of interest and an incestuous funding trail from the investors in U.S. abortion pill manufacturers Danco Laboratories and GenBioPro, whose own dollars have been funneled to various groups behind clinical trials and studies of the abortion pill, which claim it is both safe and effective.
In some instances, these studies have problematically been authored by people on the payroll of the abortion pill’s manufacturers.
2016 and 2021 FDA changes
In 2016, the FDA chose to “eliminate the requirement that prescribers report all serious adverse events,” AHM claimed, adding that “for 2016, FDA failed to consider a major aspect of the problem: ‘the cumulative effect’ of the interrelated changes.”
AHM added, “The agency, instead, noted that abortion-drug manufacturers ‘report adverse events, including serious adverse events, to FDA’… But FDA did not explain how these far-removed manufacturers would learn about adverse events from OB/GYNs, hospitalists, and emergency-room doctors who are under no obligation to report,.”
The FDA’s 2016 changes, according to AHM, “increased the risk that more women taking abortion drugs will need emergency care” in three ways:
- The FDA “increased the gestational-age limit from seven to ten weeks” where the “‘failure rate’ climbs from roughly 2 to 7 percent, as confirmed by FDA’s label.”
- The “FDA’s removal of the Day 14 in-person follow-up visit” will “naturally result[] in more women report[ing] to the emergency room.’”
- The FDA’s decision to end the “requirement that licensed doctors prescribe and provide ongoing care to women using abortion drug” results as the FDA concedes in “OB/GYNs, OB/GYN hospitalists, and emergency room doctors like Respondents ‘who must manage the aftermath.’”
As recent as 2020, the FDA “explained that it had reviewed ‘thousands of adverse events resulting from the use of Mifeprex,’ determined that abortion drugs continue to cause ‘serious risks for up to seven percent of patients,’ and concluded that an in-office visit was ‘necessary to mitigate [those] serious risks,’” the pro-life doctors claimed.
In essence, the “FDA based its decision to remove the initial in-person visit on (1) adverse event reports and (2) published literature that the agency conceded was not adequate,” the brief claimed. “Equally problematic, FDA did not recognize that it had years before abandoned the requirement that mifepristone prescribers report nonfatal adverse events.”
“Also troubling is the agency’s reliance on studies that included safety measures like ultrasound screenings and follow-up visits, even though those safeguards were not included in the approved regimen,” the brief added.
In addition, AHM alleged, many of the studies FDA relied upon for 2021 changes showed evidence that women would likely rely on ERs to handle abortion pill complications.

“Had FDA retained the safeguards that it called ‘necessary’ and ‘minimally burdensome’ just a few years ago, it could have prevented many of these emergency events,” AHM alleged. “FDA’s 2021 action also violates the Comstock Act… That statute prohibits using ‘the mails’ to send any ‘drug … advertised or described in a manner calculated to lead another to use or apply it for producing abortion.’ 18 U.S.C. 1461. It also forbids using a ‘common carrier or interactive computer service’ to ship ‘any drug … designed, adapted, or intended for producing abortion.’”
Plaintiffs Are Harmed and Therefore Have Standing
“After full briefing and argument, the Fifth Circuit held that Respondent doctors and associations have standing. The doctors suffer harm because they have no choice but to violate ‘their conscience’ by completing elective abortions in emergency situations and ‘divert[ing] time and resources away’ from their regular obstetrics practice when women present with abortion-drug complications,” the AHM brief stated.
AHM doctors have testified to treating women who took abortion drugs with severe complications — “many who presented to the emergency room.”
“In removing crucial safeguards for the use of abortion drugs, FDA expressly counted on OB/GYN hospitalists and emergency-room doctors—like Respondents—to manage abortion-drug complications,” AHM wrote. “When faced with these emergencies, Respondents have no choice but to provide immediate treatment, even though this kind of participation in an elective abortion harms their consciences and injures them in other ways.”
They allege that the “FDA’s [own] data and the Doctors’ testimony establish that Respondents face a ‘substantial risk’ of these injuries… and FDA’s 2021 and 2016 actions increase that risk.”
According to AHM, while the FDA argues they do not have standing, the FDA relied on similar hospitals for clinical trials and published abortion pill studies. One of those was the University of California, a pro-abortion institution heavily tied to abortion pill investors.
“FDA says Respondent doctors’ harms are ‘speculat[ive]’ because Respondents do not prescribe abortion drugs… But prescribing the drugs isn’t what increases Respondents’ exposure. That harm results from FDA’s removal of the safeguards. FDA’s unlawful actions have left Respondent doctors ‘to deal with preventable emergent and life-threatening situations’” AHM wrote. “FDA’s actions have forced Respondent doctors to witness and participate in the grisly process of treating abortion-drug complications—retained fetal parts, heavy bleeding, severe infections—causing the doctors mental, emotional, and spiritual distress.”
Removing Abortion Pill Safety Regs by Design Sends Women to ER’s
Live Action News previously documented that even pro-abortion emergency room staff have warned that ER doctors may see abortion pill complications fron undiagnosed ectopic pregnancies.
AHM contends in their brief before SCOTUS that “OB/GYNs, OB/GYN hospitalists, and emergency-room physicians will often be called upon to treat abortion-drug complications,” and AHM also contends that this “is not a bug in FDA’s abortion-drug plan but part of its very design. When the agency eliminated crucial safeguards in 2021 and 2016—safeguards like the ongoing care of healthcare providers—FDA justified their removal based on the availability of emergency-care coverage.”

Live Action News has documented multiple times that today, the profiteers of the deadly abortion pill regimen are flouting the FDA’s safety regulations on the abortion pill. They prescribe the pill past FDA gestational limits, may not have the ability to properly rule out potentially deadly ectopic pregnancies as required, prescribe ‘advanced provision‘ abortion pills to women who are not even pregnant, allow pharmacists to prescribe the drug, and encourage women to lie about abortion pill complications and instead to report to emergency departments claiming they are experiencing a natural miscarriage.
‘One in 25’ Abortion Pill Clients Could End up in ER
“FDA has long recognized that emergency care is essential to handle abortion-drug complications; the agency’s own numbers say that roughly one in 25 women who take mifepristone will end up in the emergency room; and Respondent doctors have testified to routinely treating women suffering abortion drug harm,” the AHM brief claimed. “Factor in that hundreds of thousands of women take abortion drugs each year in the United States, and that FDA’s own data indicates that tens of thousands of them go to the emergency room, and Respondents easily establish a ‘substantial risk’ that these harms ‘will occur’ again.”
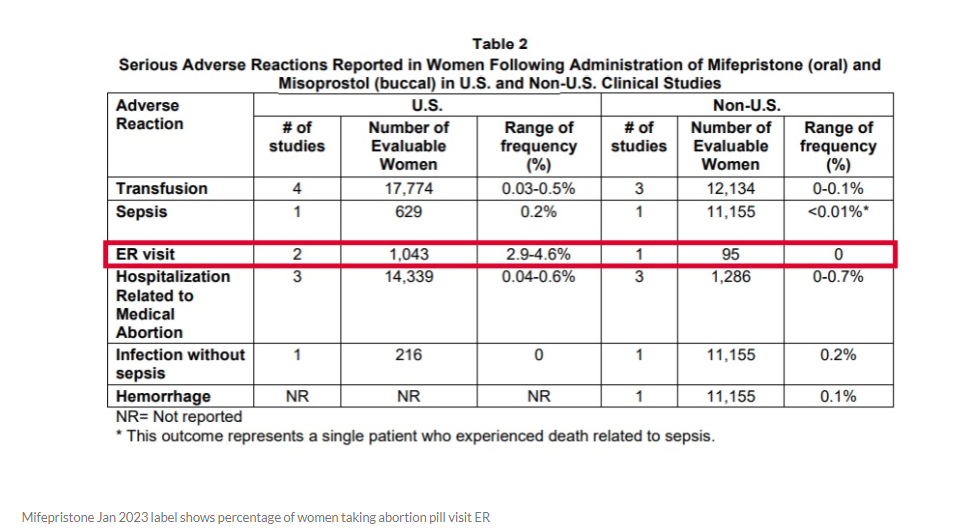
To justify that statistic, AHM wrote that the FDA has admitted that “2.9 to 4.6 percent of women who take abortion drugs end up in the emergency room.”
The Alliance Defending Freedom (ADF) who is representing AHM, directed Live Action News to mifepristone’s latest 2023 label, where in a table 2 entitled, “Serious Adverse Reactions Reported in Women Following Administration of Mifepristone (oral) and Misoprostol (buccal) in U.S. and Non-U.S. Clinical Studies,” the 2.9% – 4.6% ER statistic is located.

Black Box Warning and Medication Guide
“FDA’s current label for mifepristone continues to require a Black Box warning because the drug can cause ‘[s]erious and sometimes fatal infections and bleeding’… It also directs women to emergency rooms if one of many adverse complications arise,” AHM wrote.
The doctors added:
On that label, FDA estimates that 2.9 to 4.6 percent of women will visit the emergency room after taking mifepristone… And FDA’s medication guide acknowledges that as many as 7 percent of women will need surgery after taking mifepristone ‘to stop bleeding’ or to complete the abortion…
The label also warns that a prescriber must ‘[e]xclude [ectopic pregnancy] before treatment,’…’because some of the expected symptoms experienced with a medical abortion (abdominal pain, uterine bleeding) may be similar to those of a ruptured ectopic pregnancy.’


AHM contends that “Giving… unfettered power to FDA—an agency whose actions ‘affect every citizen’… is particularly problematic,” the brief stated. “Turning a blind eye to FDA’s patently unreasonable actions here, which jeopardize women’s health throughout the nation, would be unprecedented. No agency is above the law.”
Carole Novielli has over thirty years of research experience on abortion, Planned Parenthood and eugenics. Her work has been published by many reputable media outlets. As a woman opposed to abortion, Carole is a committed Christian who refuses to be silenced by the main stream media and the nation’s pro-abortion feminist minority.